

미국 식품의약국(FDA)이 새로운 알츠하이머 치료제가 될 것으로 관심이 쏠렸던 신약 후보에 보완 요구서한(CRL)을 요청했다.
25일 미국 바이오센추리는 FDA가 미국 일라이릴리에 CRL을 보내 알츠하이머 신약 후보물질 ‘도나네맙’에 대해 신속승인 대신 임상3상 중 안전성 데이터를 추가로 요청했다고 보도했다.
바이오센추리 또한 승인을 위해 자료를 보완하는 것이 더 유리하다는 의견이다. 아직 의료계에서 아밀로이드 베타(Aβ) 가설에 대한 의구심이 남아 있고 미국 내 공공보험(CMS) 적용이 확실하지 않은 상황에서 신속승인으로 시장에 급하게 진출하기보단 정식 품목허가를 통해 활용범위를 넓히는 것이 더 낫다는 설명이다.
Aβ 가설은 뇌에서 과다 생산·축적된 Aβ 단백질이 아밀로이드 플라크라는 끈끈한 막을 만들어 알츠하이머를 일으킨다는 가설이다.
일라이릴리는 이번 FDA 결정이 불충분한 안전성 데이터 때문이라고 설명했다. 회사 측에 따르면 FDA는 최소 12개월 동안 도나네맙으로 치료받은 환자 100명에 대한 데이터를 요구했다.
이번 신속승인 신청은 지난 임상2상(TRAILBLAZER-ALZ)에서 아밀로이드 플라크가 감소한 결과를 기반으로 한다. 일라이릴리 임상시험에서 도나네맙 치료군으로 참가한 환자는 131명이었다. 하지만 환자에서 미리 설정된 양의 플라크가 줄어든 것을 확인한 뒤 위약으로 전환했다.
지난 2021년 해외 의약저널 ‘뉴잉글랜드저널오브메디신(NEJM)’에 게재된 임상시험 결과에 따르면 투약 56주까지 환자 약 55%가 위약으로 전환하기 위한 요건을 충족했다. 도나네맙 치료 환자의 약 절반인 66명이 투약 1년안에 위약군으로 전환할 수 있는 상태에 도달한 것이다.
도나네맙은 임상2상에서 투약 6개월 뒤 환자 37.9%에서 알츠하이머 원인물질인 뇌 내 Aβ를 제거하며 효과를 입증했다.

일라이릴리는 지난 연구 결과뿐 아니라 다음 분기에 발표 예정인 임상3상(TRAILBLAZER-ALZ2) 결과에도 이와 관련한 데이터를 포함할 예정이다. 일라이릴리는 이번 임상3상 결과에 대해 “도나네맙 정식 품목허가 신청을 위한 기본자료가 될 것”이라고 말했다.
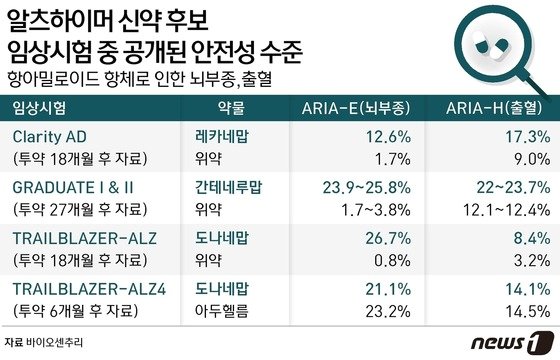
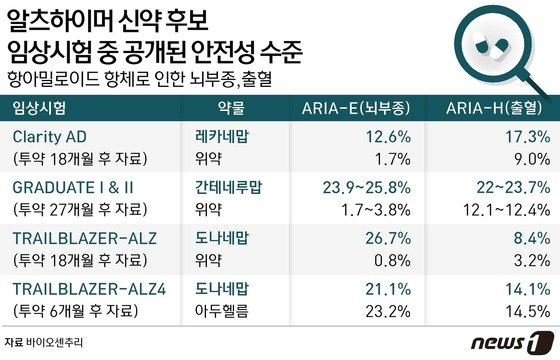
지금까지 AIRA-E(아밀로이드 관련 영상이상부종) 또는 ARIA-H(아밀로이드 관련 영상이상 미세출혈·혈철소증) 등 안전성 결과를 보면 도나네맙은 로슈의 간테네루맙과 에자이·바이오젠의 아두헬름(성분 아두카누맙) 등 다른 아밀로이드 항체와 큰 차이가 없는 수준인 것으로 나타났다.
지난 6일 FDA로부터 신속승인을 획득한 알츠하이머 신약 ‘레켐비'(성분 레카네맙)가 임상3상에서 공개했던 ARIA-E 비율은 도나네맙보다 크게 낮은 수준이었다. 현재 에자이와 바이오젠은 FDA에 레켐비 임상3상 결과를 제출해 신속승인을 정식 품목허가로 전환 신청을 한 상태이다.
바이오센추리는 또 미국 정부의 건강보험 메디케어를 운영하는 미국 보험청(CMS·메디케어·메디케이드 서비스센터)이 무작위비교임상시험(RCT)을 바탕으로 한 보험급여결정(NCD)을 적용할 예정이라 도나네맙이 신속승인 심사 대상으로 결정돼 심사가 이루어졌어도 보험혜택을 제한할 가능성이 있다고 설명했다.
CMS는 지난 2022년 초 FDA가 아두헬름 신속승인 심사를 결정하면서 RCT를 기반으로 한 ‘근거창출을 통한 보험급여'(CED)를 적용하겠다고 밝혔다. 실제로 아두헬름은 같은해 6월 승인됐지만 값비싼 가격으로 출시 첫 분기 매출이 전망치의 약 40분의 1 수준에 그쳤다.
현재 CMS는 NCD 기준을 재평가하는 중이다. 이와 관련, 바이오센추리는 레켐비가 정식 품목허가를 받으면 보험 적용 범위가 현재보다 넓어질 것으로 전망했다.


















